People with ML I can experience some or many symptoms from a wide spectrum which range from severe to very mild. The severity of ML I varies widely across 2 types which are defined by the age of the patient when the first signs and symptoms start to appear.
Type 1, symptoms can develop anywhere from childhood to young adulthood.
Type 2, symptoms can be present at birth or develop shortly after birth.
ML I type 1
People with ML I type 1 may develop symptoms anywhere from childhood to young adulthood, with most people developing symptoms during their twenties and thirties. Development and growth is normal until problems with walking or when abnormalities require medical attention. ML I type 1 does not affect intelligence or life expectancy.
Muscle twitches (myoclonus), difficulty coordinating movements (ataxia), leg tremors, and seizures are symptoms of ML I type 1. Muscle twitching worsens over time, causing difficulty sitting, standing, or walking. People with ML I type 1 eventually require wheelchair assistance. Other symptoms of ML I type 1 include the development of distinct red spots in the eyes known as cherry-red macules. People with ML I may experience loss of clarity of vision and may develop impaired colour vision and night blindness. Rapid, involuntary eye movements (nystagmus) and clouding of the cornea may also occur.
ML I type 2
ML I type 2 the more severe, early onset form, is characterised by a progressive and severe mucopolysaccharidosis-like features such as coarse facial features, enlarged organs, and developmental delay. ML type 2 is further divided into congenital, infantile, and juvenile forms.
ML I type 2 congenital
The features of congenital ML type 2 can develop before birth. This form of ML I is associated with widespread swelling before birth caused by fluid accumulation. Affected infants may also have an enlarged liver and spleen, abnormal bone development, and distinctive coarse facial features. As a result of these serious health problems infants with congenital ML type 2 usually are stillborn or die soon after birth.
ML I type 2 infantile
Infantile form shares some features with the congenital form, although the signs and symptoms are slightly less severe and begin within the first year of life. As children with infantile ML I type 2 get older, they may develop involuntary muscle jerk movements and cherry-red macules in the eyes affecting vision. Other signs and symptoms include hearing loss, overgrowth of the gums, and widely spaced teeth. Affected children may survive into childhood or adolescence.
ML I type 2 juvenile
The juvenile form has the least severe signs and symptoms. Features of this type usually appear in late childhood and may include mildly coarse facial features, mild bone abnormalities, cherry-red macules, involuntary muscle jerk movements, intellectual disability, and dark red spots on the skin. The life expectancy of people with juvenile ML I type 2 varies depending on the severity of symptoms.
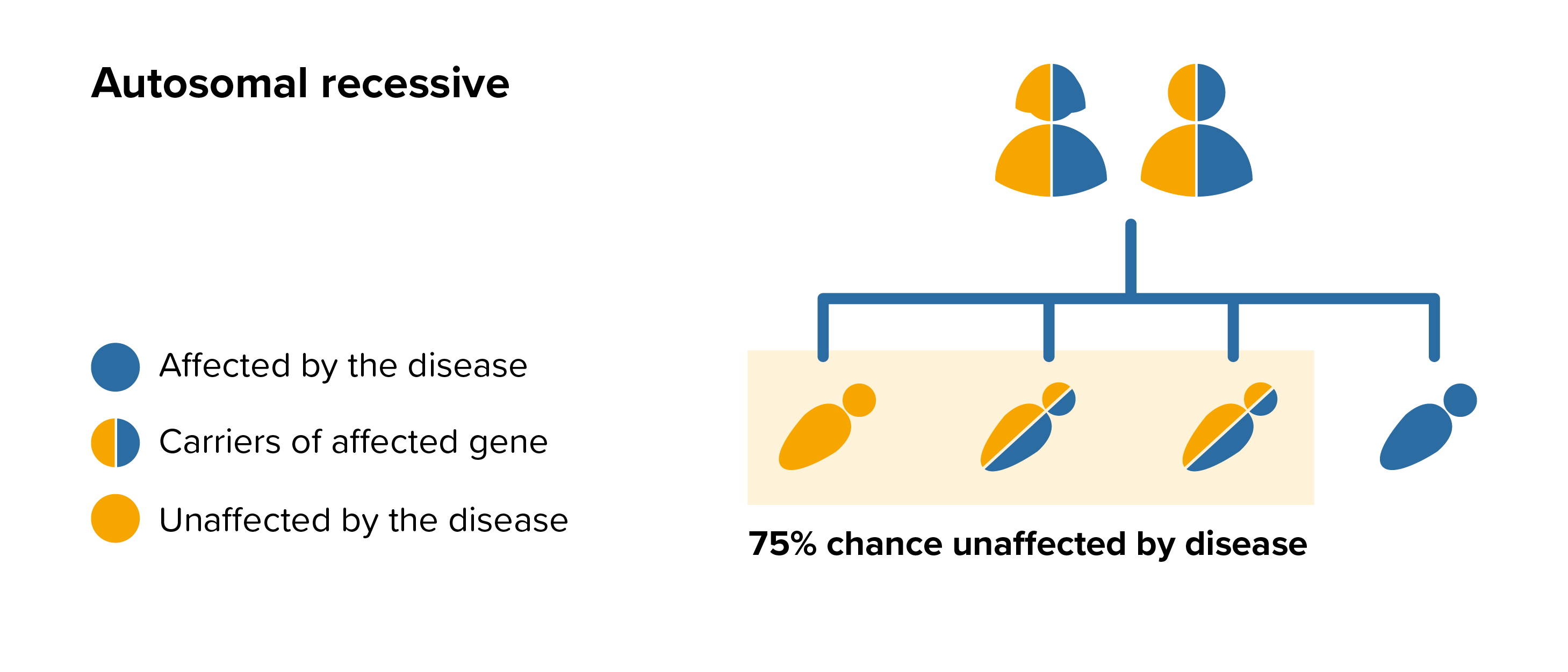 All parents of children with ML I can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by ML I, screening tests can be arranged early on during a pregnancy for those families who already have a child with ML I. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.
All parents of children with ML I can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by ML I, screening tests can be arranged early on during a pregnancy for those families who already have a child with ML I. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.




