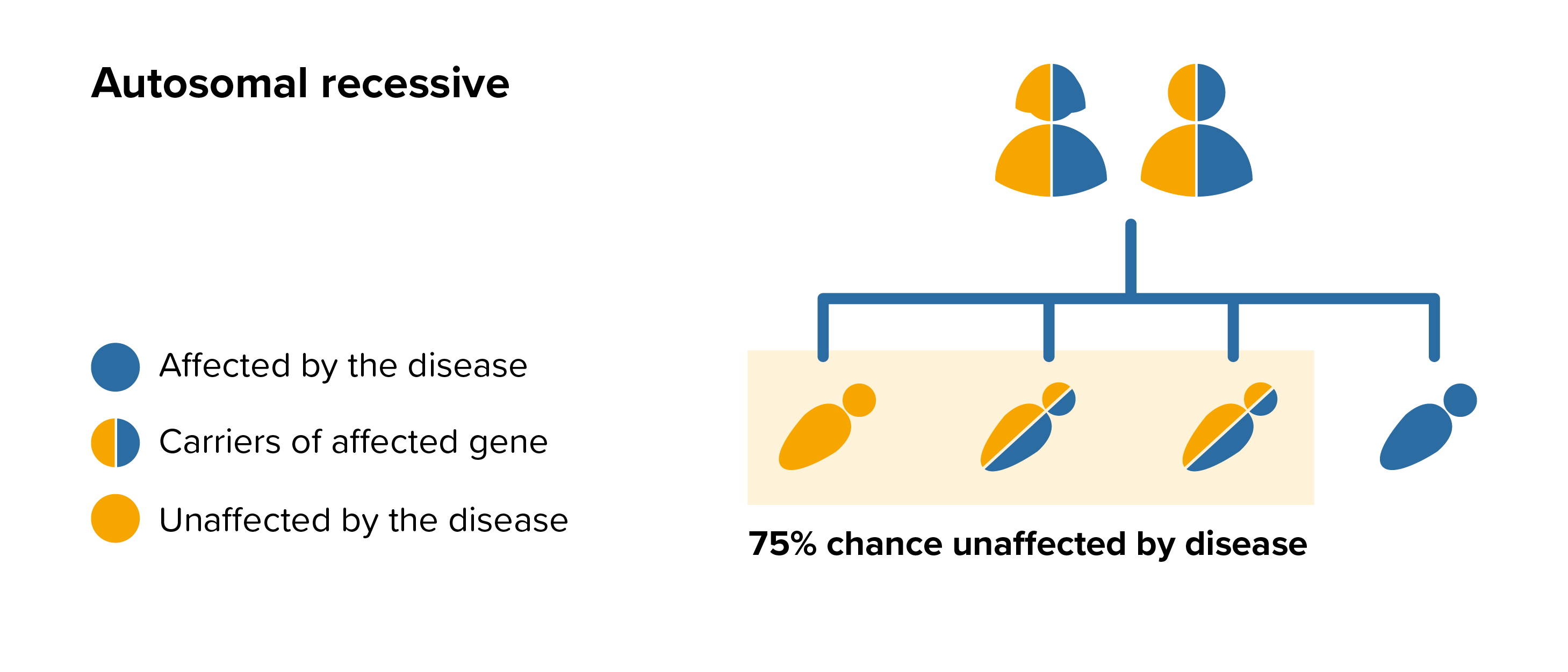
ML IV is an autosomal recessive disease this means that both parents must carry the same affected gene and each pass this same affected gene to their child.
People probably carry from 5 to 10 genes with mutations in each of their cells. Problems happen when the particular gene is dominant or when a mutation is present in both copies of a recessive gene pair. Genes are the unique set of instructions inside our bodies that make each of us an individual. They are the blueprint for our growth and development, as well as controlling how our bodies function.
Genes are carried on structures called chromosomes and it is usual to have 23 pairs. A child will inherit half of the chromosomes from the mother and the other half from the father resulting in 23 pairs. 22 of these pairs look the same in both males and females. Pair 23 are the sex chromosomes, and this is the pair that differ between females and males. The X chromosome is inherited from the mother and the Y chromosome is inherited from the father. More information about inheritance is available in our publication.
For each pregnancy the chances of a baby inheriting ML IV are completely independent of whether a previous child was affected with ML IV. With each pregnancy there is a 1 in 4 chance that the baby will be affected by ML IV.
 All parents of children with ML IV can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by ML IV, screening tests can be arranged early on during a pregnancy for those families who already have a child with ML IV. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.
All parents of children with ML IV can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by ML IV, screening tests can be arranged early on during a pregnancy for those families who already have a child with ML IV. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.
Amniocentesis and chorionic villus sampling are both available during the pregnancy to find out if the baby is affected by ML IV.




