
Want to know more about Fabry? Watch our animated guide.
On this page you can find our animated guide, general information about the condition, latest news, updates and stories and a list of relevant resources and events.
Understanding the condition
What is Fabry?
Fabry disease is closely related to mucopolysaccharidoses and is one of the lysosomal storage diseases. It was first described in 1898 by William Anderson and Johannes Fabry and is also referred to by some as Anderson–Fabry disease.
What causes Fabry?
In the course of normal life there is a continuous recycling process in the body which consists of building new materials and breaking down old ones ready for disposal. This activity takes place in a special part of the body’s cells called the lysosome. This process requires a series of biochemical tools called enzymes. The enzyme alpha-galactosidase A (alpha-GAL) is essential in breaking down the fatty acid globotriaosylceramide (GL3).
People with Fabry cannot make enough of alpha-GAL, without enough levels of the enzyme the normal functioning of vital organs is affected. When GL3 is not completely broken down it builds up within the cells of the body causing progressive damage. Organs such as kidney, heart and brain eventually start to deteriorate, and severe or life-threatening complications can arise. Babies may show little sign of the disease but as more and more cells become damaged by an accumulation of these waste products, symptoms start to appear.
Frequently asked questions
People probably carry from 5 to 10 genes with mutations in each of their cells. Genes are the unique set of instructions inside our bodies that make each of us an individual. They are the blueprint for our growth and development, as well as controlling how our bodies function. Genes are carried on structures called chromosomes and it is usual to have 23 pairs.
A child will inherit half of the chromosomes from the mother and the other half from the father resulting in 23 pairs. 22 of these pairs look the same in both males and females. Pair 23 are the sex chromosomes, and this is the pair that differ between females and males. The X chromosome is inherited from the mother and the Y chromosome is inherited from the father. More information about inheritance is available in our publication.
The inheritance pattern for Fabry is called X linked semi-dominant inheritance. Males only have one X chromosome, if it contains the faulty gene which cannot make enough enzyme alpha-GAL they are more likely to develop Fabry symptoms. Males can only pass the faulty gene to the daughters, so the daughters will be carriers of the disease, sons will not be affected by Fabry.
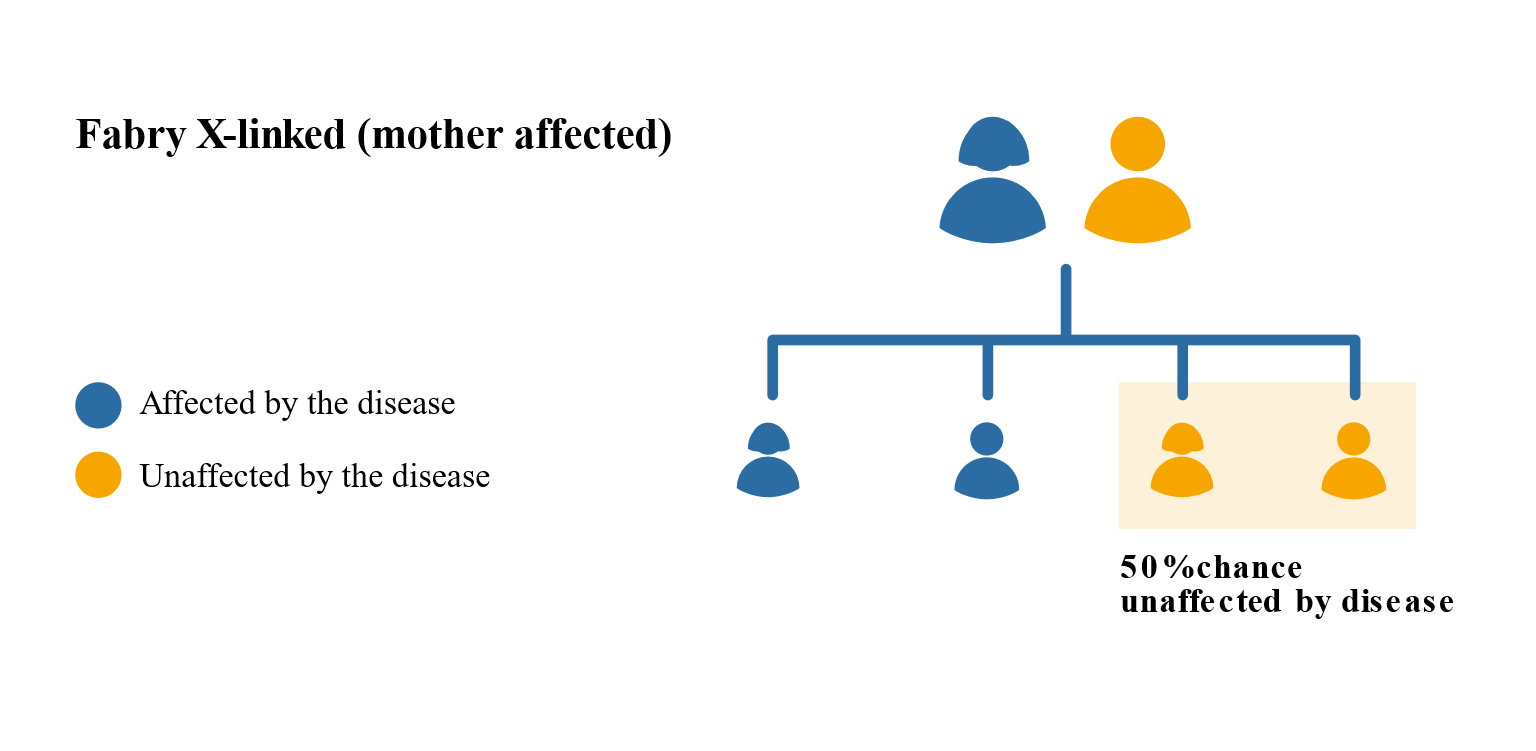
 Females may have less severe Fabry symptoms than males but this is not always the case. This is because females have two X chromosomes, one of which will be active and one inactive. It is a matter of chance which chromosome is active or inactive in a particular cell. If the X chromosome with the faulty gene is active then a female is likely to show many of the classic features of Fabry. There is a 1 in 2 chance that that faulty gene will be passed on to the children, regardless of whether they are male or female.
Females may have less severe Fabry symptoms than males but this is not always the case. This is because females have two X chromosomes, one of which will be active and one inactive. It is a matter of chance which chromosome is active or inactive in a particular cell. If the X chromosome with the faulty gene is active then a female is likely to show many of the classic features of Fabry. There is a 1 in 2 chance that that faulty gene will be passed on to the children, regardless of whether they are male or female.
 All parents of children with Fabry can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by Fabry, screening tests can be arranged early on during a pregnancy for those families who already have a child with Fabry. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. Amniocentesis and chorionic villus sampling are both available during the pregnancy to find out if the baby is affected by Fabry.
All parents of children with Fabry can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by Fabry, screening tests can be arranged early on during a pregnancy for those families who already have a child with Fabry. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. Amniocentesis and chorionic villus sampling are both available during the pregnancy to find out if the baby is affected by Fabry.
It is also possible to have Pre-implantation genetic diagnosis (PGD) screening to avoid passing Fabry to the baby. PGD is an assisted fertility treatment that involves checking the chromosomes of embryos before they are transferred in the womb using IVF techniques.
It is estimated that nearly 6% of the UK population (around 3.5million people) will be affected by a rare disease at some point in their lives. A single rare disease may affect up to about 30,000 people however the vast majority of rare diseases affect far fewer than this.
It has been estimated that Fabry affects between 1:40,000 people, in Caucasian males it is around 1:17,000. Fabry is seen across all ethnic groups, including African Americans and in persons of Hispanic or Asian descent but the population-specific incidence rates are unknown.
Disease progression is influenced by the sex of the person (male or female) and whether the disease presents as either non-classical (mild form) or classical (severe form). People with the classic form often demonstrate the full spectrum of symptoms.
Early onset begins in childhood or adolescence and include cloudiness of the front part of the eye (cataracts), ringing in the ears (tinnitus) and hearing loss, clusters of small dark red spots on the skin (angiokeratoma), pain in the extremities, and decreased ability to sweat (hypohidrosis). In addition, they may have chronic abdominal pain and diarrhoea. Fabry disease also involves potentially life-threatening complications such as progressive kidney damage, heart attack, and stroke.
Other symptoms can include tiredness that is not relieved by rest or sleep, intolerance to heat or cold and a feeling of burning in the hands and feet. Sometime people can feel dizzy (vertigo) and signs of depression are present. People with Fabry often have difficulties in managing weight, feeling sick or being sick and pain or bloating after eating, difficulty managing weight and feeling full after eating a small amount of food.
Some people with the milder forms of the disease that appear later in life and affect only the heart or kidneys. Death, as a result of renal failure, heart failure, or strokes, commonly occurs by the fourth or fifth decade of life.
Chaperone therapy
Chaperones are small molecules that assist enzymes in becoming functional by helping them take the correct shape and stay stable. With chaperone therapy, some faulty forms of alpha-GAL enzyme can be corrected and delivered to the lysosomes so that the excess GL3 can be broken down.
The name for the chaperone therapy treatment in Fabry is migalastat and the brand name is Galafold®. Galafold® was licensed as the first oral chaperone therapy in Fabry and has been available in the UK since 2016 for adults and adolescents 16 years of age and older. Patients must have a confirmed diagnosis of Fabry and the mutation in the galactosidase alpha (GLA) gene that produces an alpha-GAL enzyme that responds to the treatment.
Galafold® is an oral capsule taken every other day. More information about Galafold® can be found at www.galafold.co.uk and a European version of the patient information leaflet is here. Further information on this treatment is available from the electronic medicines compendium.
Enzyme Replacement Therapy (ERT)
For people with Fabry ERT is a long-term therapy whereby the missing or deficient enzyme is given via an intravenous infusion. Currently Fabrazyme ® (agalsidase beta) and Replagal ® (agalsidase alpha) are both ERTs for the treatment of Fabry and both are approved in the UK. ERT helps to normalise kidney and heart function, and blood supply to the brain.
Fabrazyme ® is an infusion administered every other week at home for patients aged 8 years and older. More information about Fabrazyme® can be found at www.fabrazyme.com and a UK version of the patient information leaflet is here. Further information on this treatment is available from the electronic medicines compendium.
Replagal® is an infusion administered every other week over 40 minutes and is usually administered at home for patients aged 7 years and older. More information about Replagal® can be found at www.shire.com and a UK version of the patient information leaflet is here. Further information on this treatment is available from the electronic medicines compendium.
For an up-to-date list of current UK based trials taking place visit Be Part of Research (resource provided by the National Institute for Health Research). For an international search visit Clinical Trials (resource provided by the U.S. National Library of Medicine).
This resource provides information on trial status including recruiting, completed or withdrawn and worldwide trial locations. To find out more about past or current trials speak to your doctor and learn about the risks and potential benefits.
The MPS Society is the only UK charity at the forefront of supporting people and families affected by MPS and related diseases. Our extensive support services offer you a wide range of support and resources.
The team can advise and sign post you to adequate needs-led support and services in your local area as well as social care, home adaptions, education and much more.
The support team can visit you in your home and provide you with vital support.
Get involved and support us in the community, volunteer or support fundraising; we are a small charity but with your support we can continue to offer a highly valued and essential service.
Latest resources

Review: Fabry Education and Resilience Project 2025
Discover the core themes our Fabry community discussed throughout the Fabry Education and Resilience Project and access helpful resources to navigate life with Fabry.

Understanding Fabry disease - information for parents and families
We know that being diagnosed with a rare disease is life-changing and you can struggle to come to terms with it. Therefore, we have...

My Fabry treatment: a guide to empower and inform Fabry patients on their treatment journey
This guide is for patients with Fabry disease. It explains the structure of care you may receive at your specialist centre.

Living well with Fabry: a shared decision-making toolkit
This toolkit has been developed in collaboration with people living with Fabry and Fabry specialists and can help you talk about some of the symptoms that people have told us impact them the most.

Living with Fabry disease - David Moreno-Martinez
Dr Moreno-Martinez talks about living with Fabry disease and what it means to navigate a rare genetic condition at Fabry Matters Conference 2024.

Family communication - Alison Wilson
Alison Wilson, Senior Support & Advocacy Officer at the MPS Society, explains how Fabry is inherited and highlights why family communication matters.
Fabry findings

Fabry Findings - Issue No. 5 - Autumn 2021
Findings from research conducted with the Fabry community by Rare Disease Research Partners on behalf of the Fabry International Network.

Fabry Findings - Issue No. 4 - Autumn 2020
Findings from research conducted with the Fabry community by Rare Disease Research Partners on behalf of the Fabry International Network.

Fabry Findings - Issue No. 3 - Spring 2020
Findings from research conducted with the Fabry community by Rare Disease Research Partners on behalf of the Fabry International Network.

Fabry Findings - Issue No. 2 - Winter 2019
Findings from research conducted with the Fabry community by Rare Disease Research Partners on behalf of the Fabry International Network.
Latest news and blogs

Condition
Chiesi receives MHRA approval for additional dosing regimen of Elfabrio
We are pleased to share that Chiesi has received MHRA approval for an additional dosing regimen for pegunigalsidase alfa in adults stable with an enzyme replacement therapy (ERT).
14/05/2026

Condition
The importance of clinical guidelines
Regardless of location and specialist teams, clinical guidelines exist to ensure the same high standard of care. Learn more about our involvement in their development and see which conditions have so far been published.
21/08/2025

Blog
Wellbeing Wednesday
We know that living with a rare condition is tough. Some days are good, but some days aren’t! Our goal is to equip you with a ‘bag of tricks’ that you can pick and choose from when you need an extra pick-me-up.
27/05/2025
Your stories

Story
Starting treatment
Dave has recorded an insightful video to offer an insight into his first ever Fabrazyme infusion.
18/09/2025
Story
Chat with Liz for International Fabry Women's Day
The first Saturday in April is International Fabry Women's Day and we had a lovely chat with Liz who has had a very recent diagnosis with Fabry.
05/04/2025

Story
Conclusion of an epic challenge
After 12 months, Steven Gill completes his heroic effort of challenging himself across 59 events to raise funds for the MPS Society.
04/01/2024

Need someone to talk to?
Our support includes an active listening service and telephone helpline.