
What is MPS III?
MPS III, known as Sanfilippo disease, is one of the mucopolysaccharide storage diseases. MPS III was first identified by Dr Sanfilippo in 1963 and includes four different types: A, B, C and D.
Mucopolysaccharides are long chains of sugar molecules used in the building of bones, cartilage, skin, tendons and many other tissues in the body. In the course of normal life there is a continuous recycling process of building new mucopolysaccharides and breaking down old ones, which requires special biochemical tools called enzymes.
People with MPS III are missing, are low in or have altered enzymes from one of four specific types, which means they can’t break down the mucopolysaccharide heparan sulphate.
When heparan sulphate is not completely broken down it remains stored in the body, and this causes the symptoms that people with MPS III experience.
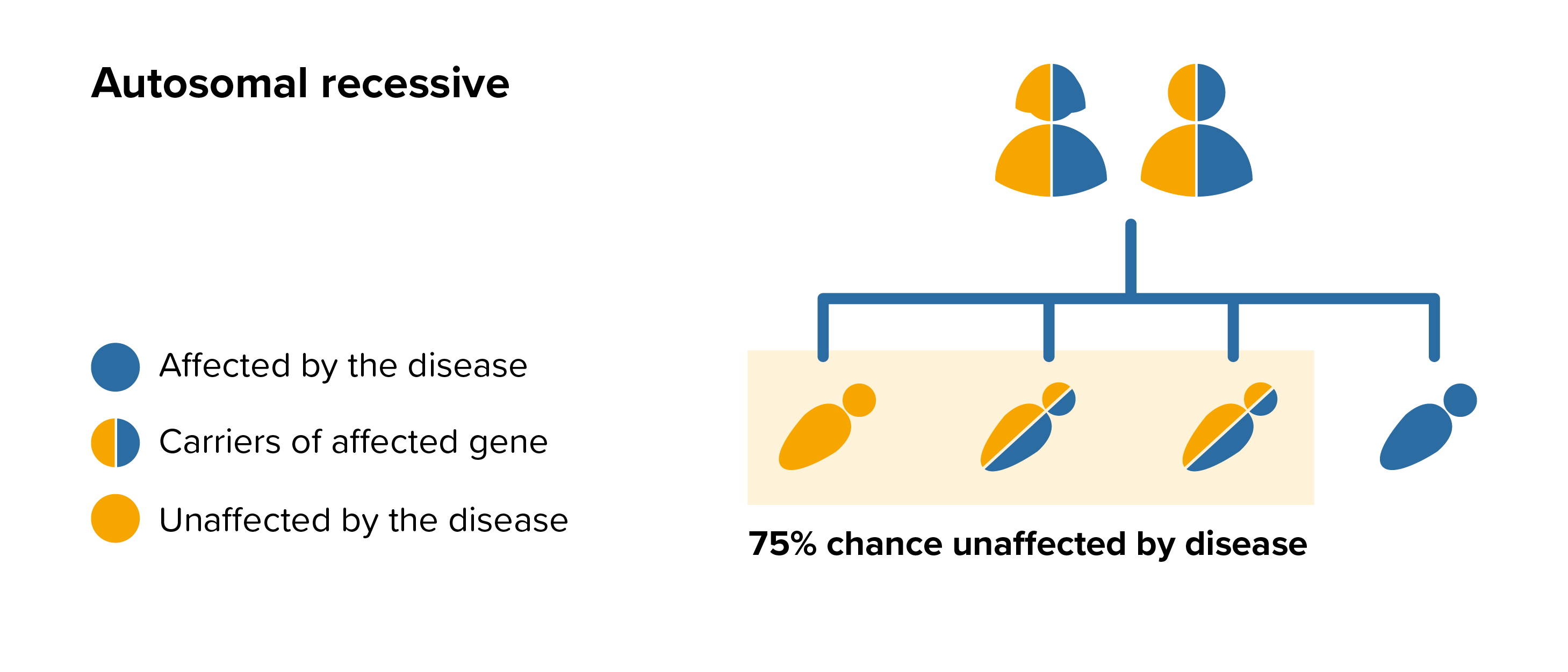 Genetic counselling
Genetic counselling









